索鎏敏團隊:電場增強陽離子疏水分子篩降低寬電位水系電解液濃度閾值
來源:原創 | 2022年09月27日

高濃度Water-in-Salt電解液拓寬了了水系電解液的窗口,使水系鋰離子電池能夠獲得大于2V的高電壓。但是,帶有高鹽濃度的水系鋰離子電池會存在成本高、動力學性能差、低溫性能差等問題,阻礙了水系鋰離子電池商業化的進程。
近日,中國科學院物理研究所的索鎏敏、華中科技大學的馮光團隊發現,只需要在水系電解液中加入少量的含有疏水陽離子的添加劑,即可將水系電解液所需的鋰鹽減少一半,而且擁有與高濃度水系電解液相近的電化學窗口(3.23V)。這些現象歸因于所添加的陽離子能夠形成電場增強的陽離子分子篩,從而阻止水分子在負極界面發生析氫反應。同時,降低水系電解液的濃度能為水系鋰離子電池帶來諸多好處,包括低成本、低粘度、高的離子電導率等。該文章發表在國際頂級期刊Advanced Materials,周安行和張錦凱為本文的第一作者。
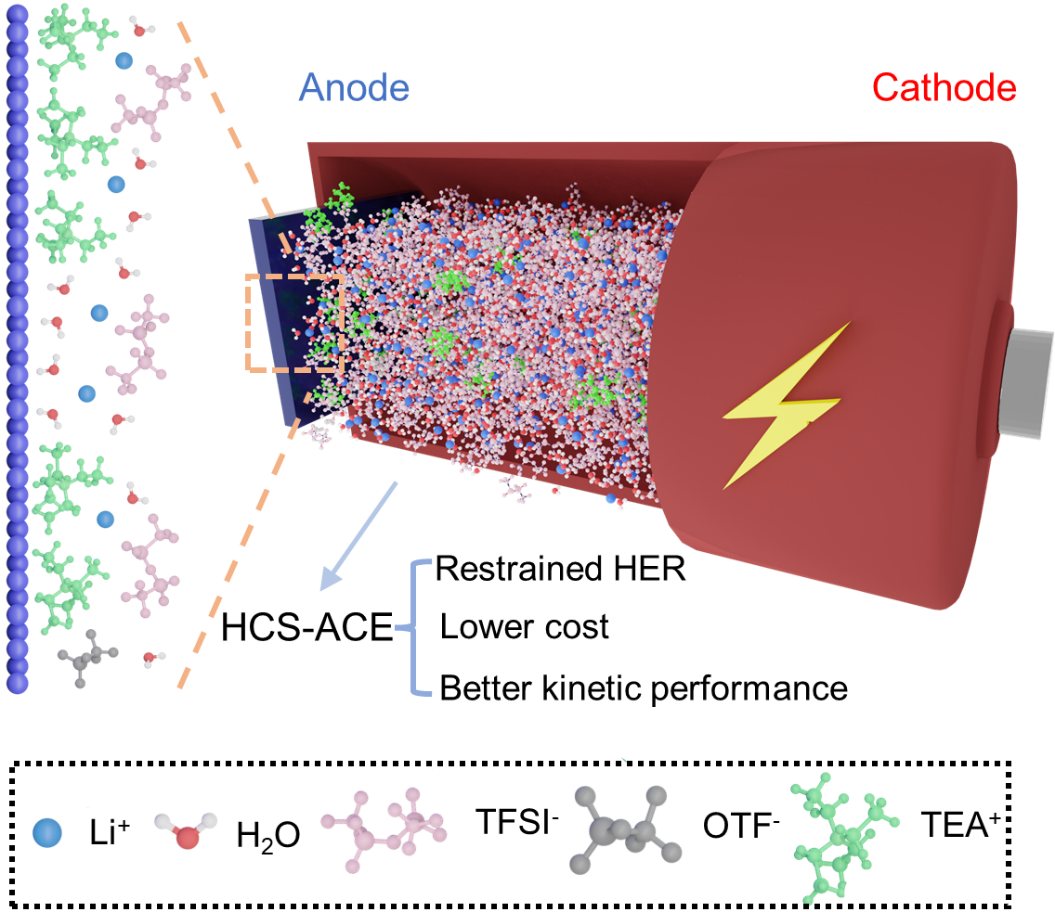
1.在負極的界面上構筑疏水陽離子分子篩
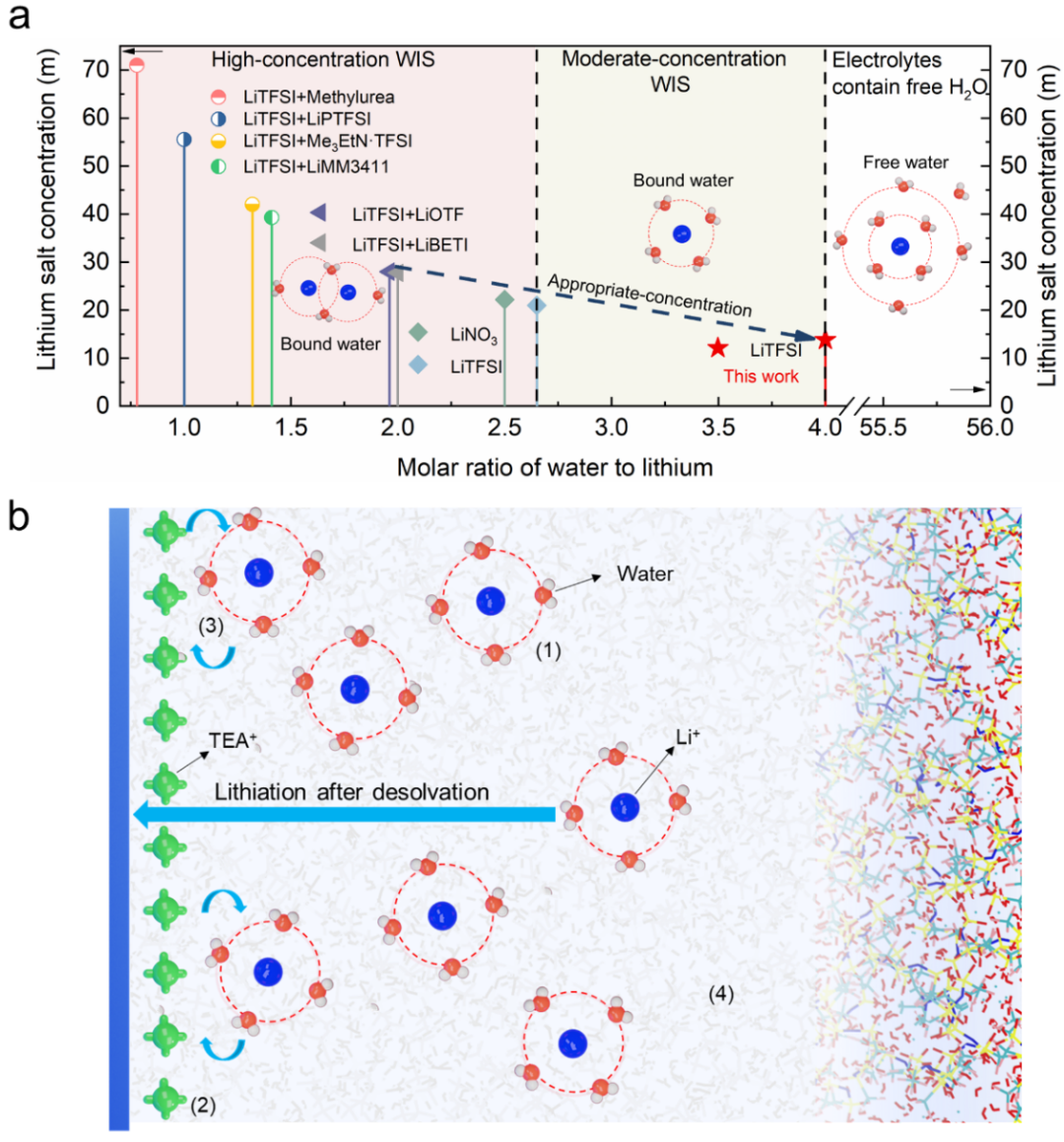
圖1.疏水陽離子界面分子篩的建立。a)已報到的文獻中各種電解液鋰鹽的濃度和水與鋰的摩爾比。b)疏水陽離子分子篩的示意圖。圖中的標簽為:(1)為結合水,(2)為疏水陽離子分子篩,(3)為抑制析氫反應,(4)為合適的電解液濃度。
在理論上,鋰離子溶劑化第一層殼層中的水分子數量是4,如果僅僅考慮通過鋰離子的溶劑化作用降低水分子的活性,那么鋰與水分子的摩爾比為1:4即可,此時對應的鋰鹽濃度為13.8m,我們定義13.8m LiTFSI為合理濃度電解液(ACE,圖1a)。當負極嵌鋰時,溶劑化的鋰離子能夠將水分子帶到負極的界面區域,從而引起析氫反應。為了盡可能的減少水分子接觸到負極界面從而發生反應,我們往ACE中引入了一定量的三氟甲磺酸四乙銨(TEAOTF),在電場的作用下,TEA+大陽離子會聚集在負極的界面,形成疏水陽離子分子篩(HCS),并且由于TEA+陽離子具有較大的半徑和較弱的水合能力,能夠阻擋水分子在負極界面發生反應,從而創造一個難以發生析氫反應的化學環境(圖1b)。
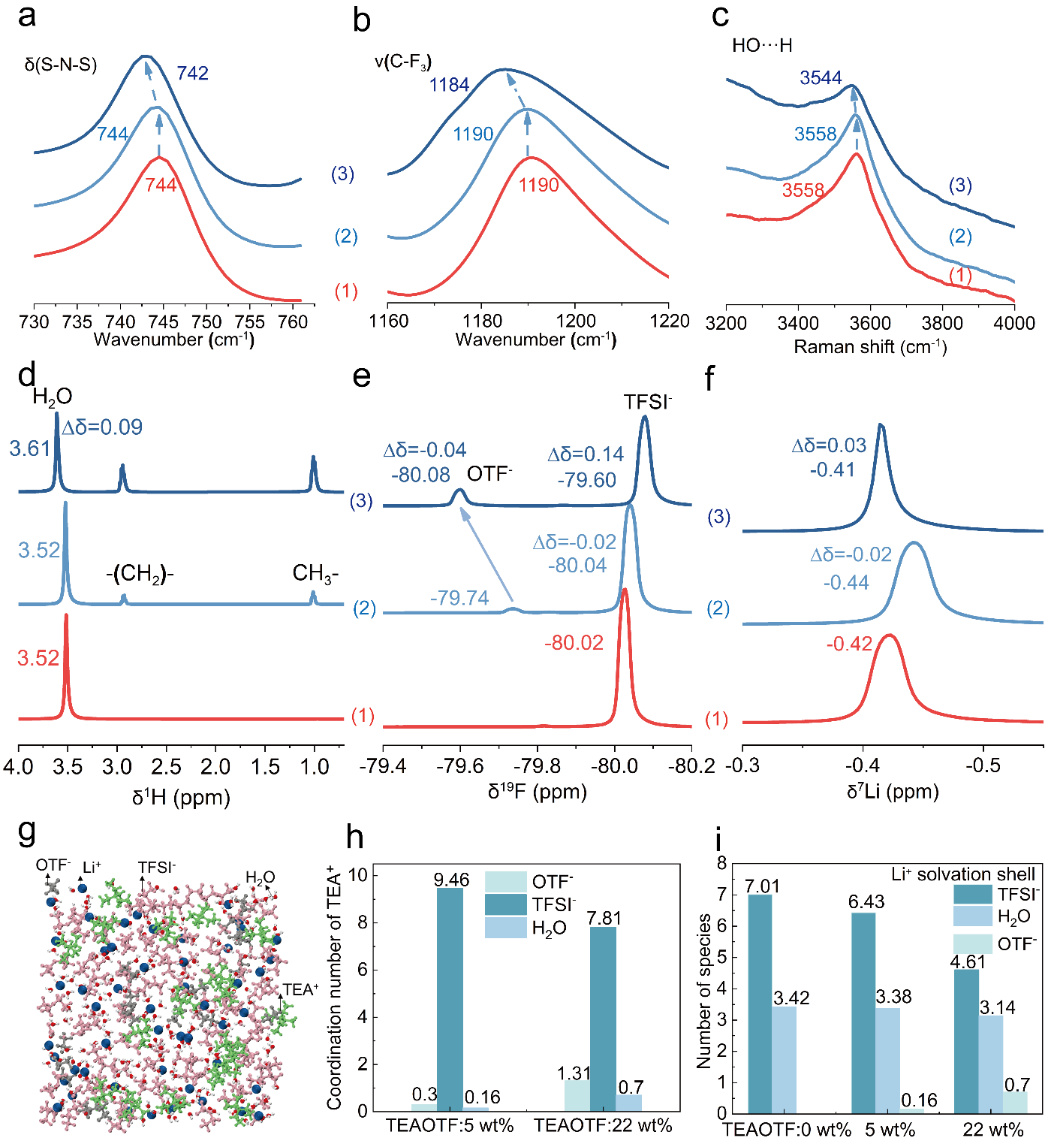
圖2.在不同TEAOTF添加劑含量下ACE電解液中陰陽離子的相互作用。在ACE中不同添加劑TEAOTF的添加量(0wt.% (1),5 wt% (2),22 wt.%(3))下a)紅外光譜中的S-N-S振動模式,b) C-F3對稱伸縮模式,c)水分子的O-H伸縮模式。在ACE中不同添加劑TEAOTF的添加量(0wt.% (1),5 wt% (2),22 wt.%(3))下的核磁共振化學位移,d)1H,e)19F,f)7Li。g)當TEAOTF添加劑的含量為5wt%時,分子動力學模擬的快照。h)由分子動力學模擬得到的TEA+大陽離子周圍其他物質的配位數。i)鋰離子殼層中其他物質的的配位數。
為了確定添加劑TEAOTF的最佳使用量,進行了紅外、拉曼、核磁共振、分子動力學模擬等表征。可以明顯的發現,再往ACE中添加5wt%的TEAOTF時,紅外光譜中的S-N-S振動模式,C-F3的堆成伸縮模式,以及拉曼光譜中水分子的O-H伸縮模式基本保持不變,但是當添加劑TEAOTF的量增加到22wt%時,上述光譜出現了明顯的紅移,說明過多TEAOTF的加入可能會影響Li+與水的溶劑化結構,減弱水和鹽之間的相互作用力(圖2a-c)。這樣的結果能夠通過核磁共振得到證實。在1H譜中,水分子的峰在添加了5wt%的TEAOTF幾乎保持不變,但是當添加劑含量提高到22wt%時,水分子的峰出現了低場的化學位移,顯示出一個弱的電子云密度(圖2d)。在1F中,OTF-和TFSI-陰離子的峰出現了兩種相反的趨勢,TFSI-中F的峰出現了高場的化學位移,OTF-中的F出現了低場化學位移,說明TFSI-中F周圍的電子云密度增加,OTF-中F周圍的電子云密度降低,這樣的結果說明了TEA+-TFSI-和TEA+-OTF-之間相互作用的趨勢是:TEA+-TFSI-> TEA+-OTF-(圖2e)。在7Li譜中,當TEAOTF添加劑的量為5wt%時,Li的信號出現了一個高場偏移,說明了鋰離子周圍出現了強的離子屏蔽,當TEAOTF的量增加到22wt%時,又出現了一個低場偏移,說明了過多了TEAOTF的加入會影響Li與水的溶劑化結構,可能會增加水的活性(圖2f)。
紅外、拉曼、核磁的結果能夠通過分子動力學模擬得到更好的理解(圖2g)。當TEAOTF的加入量為5wt%時,TEA+周圍TFSI-的量會遠大于OTF-的量,印證了TEA+-TFSI-和TEA+-OTF-之間相互作用的趨勢是:TEA+-TFSI-> TEA+-OTF-(圖2h)。并且,當TEAOTF的加入量增加時,Li+周圍水分子的含量會出現一個下降,證明了TEAOTF的量增多會使部分結合水變成自由水(圖2i)。
因此,我們認為5wt% TEAOTF的加入能夠在Li+,水分子和各種陰離子之間達到很好的平衡,所以我們將加入5wt% TEAOTF的ACE定義為HCS-ACE。
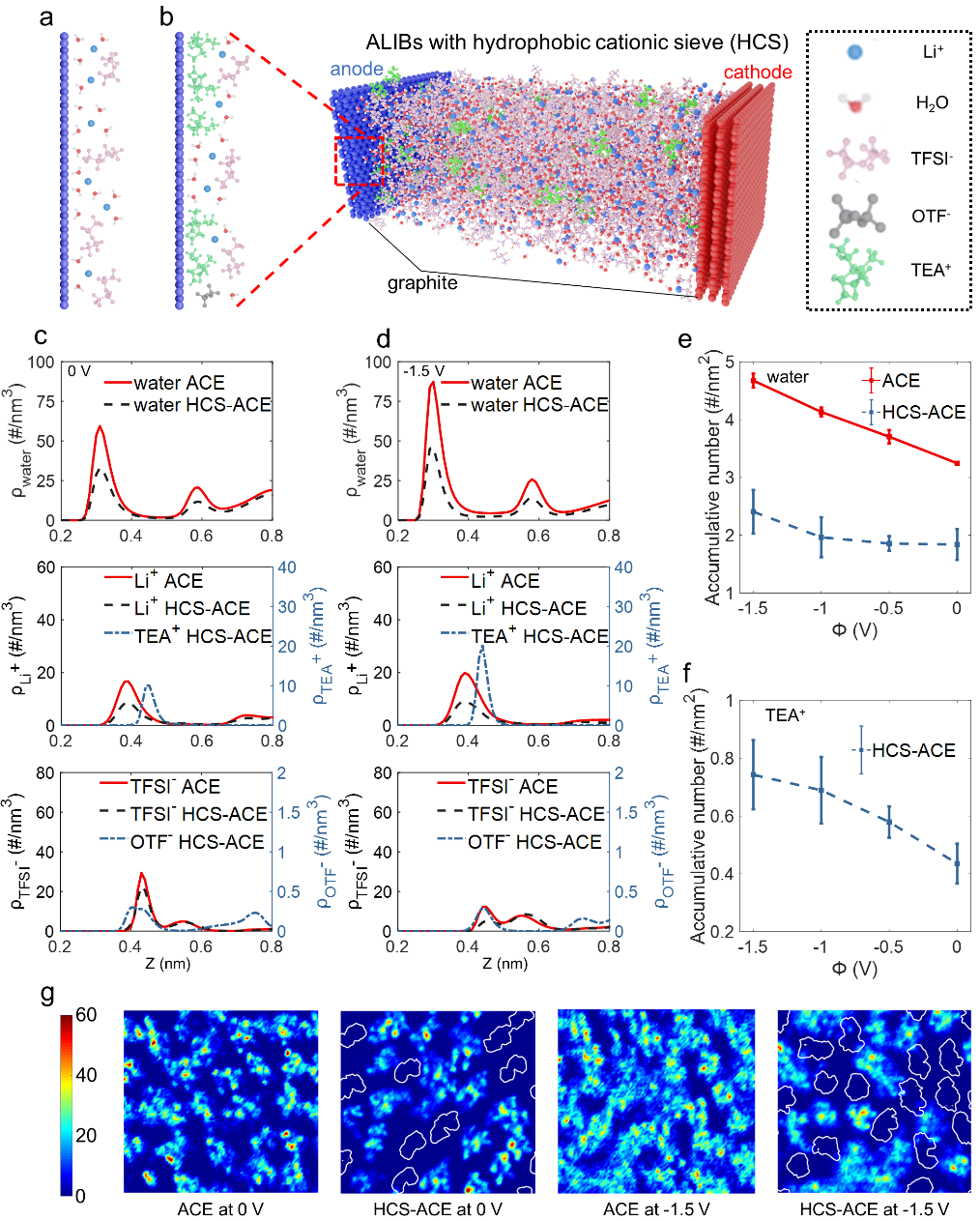
圖3負極的界面結構。a)沒有HCS的負極界面結構。b)含有HCS的分子動力學模擬結果,以及含有HCS的負極界面結構。在0V c)和-1.5V d)時水分子(頂部),陽離子(中間),陰離子(底部)的數密度圖。e)在HCS-ACE和ACE兩個系統中,負極界面水分子電吸附的情況。f)在HCS-ACE系統中TEA+陽離子電吸附的情況。g)在負極界面水分子二維分布的情況,白色輪廓代表TEA+的位置。
為進一步研究添加劑對HER的影響,我們使用等電勢分子動力學模擬技術對碳包覆TiO2負極-電解液界面進行了模擬(圖3a-b)。加入TEAOTF后,在不同電極極化下,水的第一個吸附峰都明顯下降,Li+和TFSI-也呈現出相同變化(圖3c-d)。為了定量地表現界面吸附的情況,作者比較了加入TEAOTF前后各組分在界面區域內的平均密度。加入TEAOTF后,界面水減少了40­%­–50%(圖3e)。界面自由水占界面水的10%–15%,加入TEAOTF后減少了20%–46%(支持性數據S14)。盡管只加入了少量TEAOTF,在0V時仍能觀察到TEA+的吸附峰,此時TEA+與電極的范德華相互作用能較強,與此對應的,TEA+吸附峰和低于體相區域的自由能勢阱位置一致。模擬觀察到TEA+的吸附導致的界面結構變化使得零電荷電勢正向偏移,和實驗測量結果一致。隨著負極極化增大,由于電極和TEA+間的靜電相互作用增強,TEA+自由能勢阱的最低值降低,導致了TEA+在負極的聚集,形成了隨著電場增強的HCS(圖3f)。作者進一步計算了各組分在界面區域的二維數密度分布(圖3g)。水的位置和Li+鄰近,和大部分界面水為結合水相符,而極少水處于較疏水的TFSI-占據區域。TEAOTF加入后,電極界面區域大部分空間被HCS占據,同時水占據的空間減少。帶有四個乙基支鏈的TEA+,和水的相互作用在所有組分中最弱,且其尺寸能在負極界面實現強的位阻效應,從而能在負極界面減少水的賦存。電極電勢越負,HCS占據空間越大,水占據空間越小。因此,添加TEA+修飾了界面結構,使得水更難觸及到電極表面,導致HER更難發生。
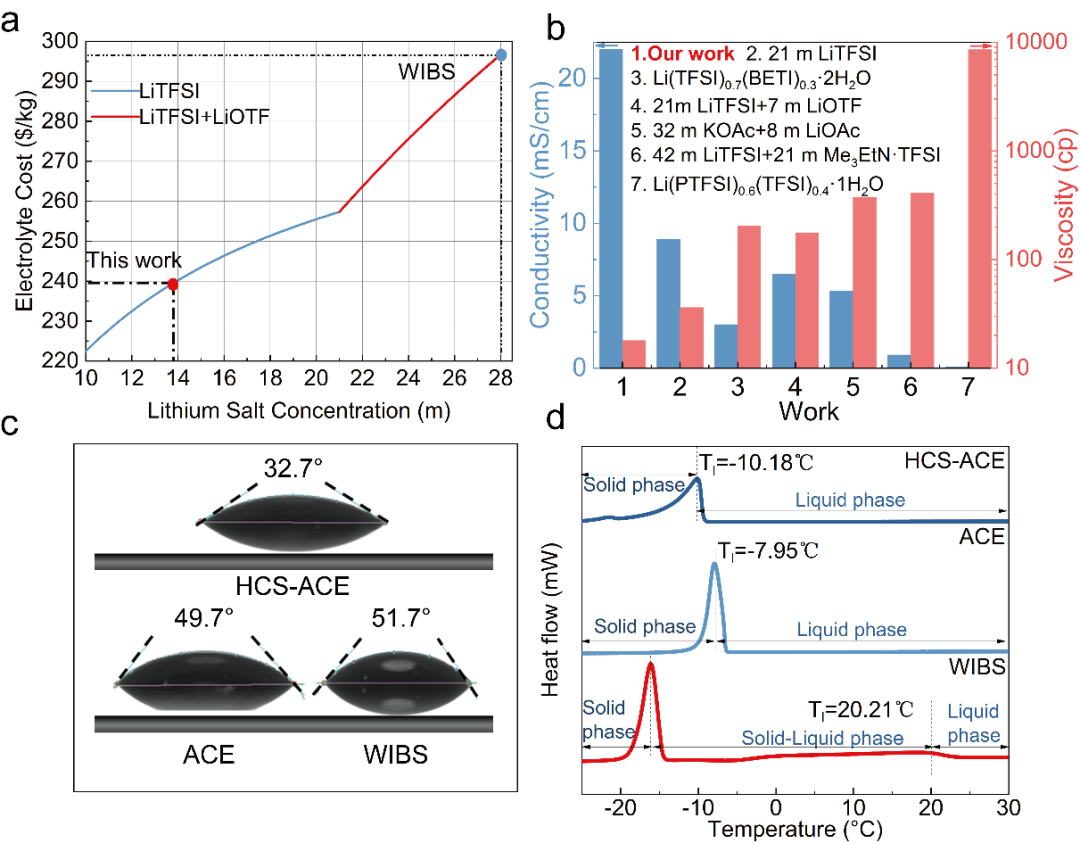
圖4.HCS-ACE的物理化學性能。a)HCS-ACE的成本分析,與WIBS進行對比。b)HCS-ACE與已發表的其他電解液進行粘度電導率的對比。c)HCS-ACE與ACE和WIBS接觸角的對比。d)HCS-ACE,ACE,WIBS熱穩定性對比。
前期的工作我們報到過雙鹽混合的WIBS(21m LiTFSI+7 mLiOTF),但是此電解液由于具有較高的鋰鹽濃度,從而具有較高的成本,本工作中,由于鋰鹽濃度的降低,帶來了比WIBS更低的成本(圖4a),并且由于鋰鹽濃度的降低,帶來了諸多好處,例如目前在WIS電解液體系中最高的離子電導率和最低的粘度(圖4b),并且由于TEAOTF屬于一種表面添加劑,能夠減少水的界面張力,所以在三種電解液HCS-ACE,ACE和WIBS中,具有最好的浸潤性能(圖4c)。并且HCS-ACE具有較低的固液轉變溫度(圖4d),在電池的低溫性能上也會具有優勢。
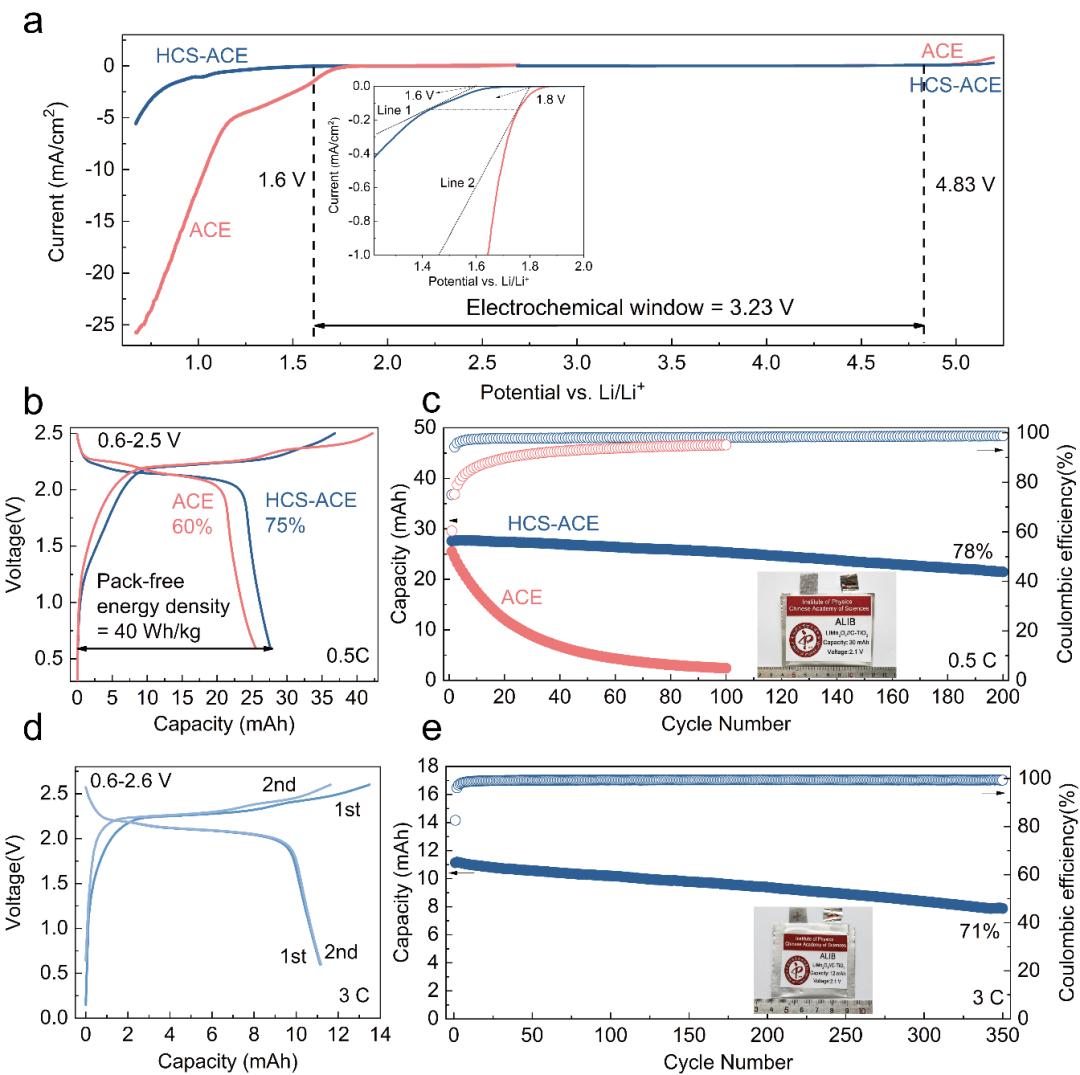
圖5.ACE和HCS-ACE的電化學性能。a)HCS-ACE與ACE的電化學窗口對比。b)不同電解液下mAh級別軟包電池的首周充放電曲線,庫倫效率以及能量密度(HCS-ACE)。c)mAh級別的軟包電池在ACE和HCS-ACE電解液下的循環性能。d)mAh級別的軟包電池在HCS-ACE下3C的循環曲線。e)mAh級別的軟包電池在3C倍率下的循環性能。
我們測量了HCS-ACE和ACE的電化學窗口(圖5a),可以看出HCS-ACE的電化學窗口為3.23V,能夠與傳統的28m的WIBS進行比較。并且明顯的是在析氫側,相對于ACE,HCS-ACE的析氫窗口拓寬了0.2V,即使是在極端的1V vs. Li/Li+,HCS-ACE的析氫電流密度也要比ACE小一個數量級,能夠明顯的顯示出HCS對于抑制析氫反應的能力。同時,我們還對兩種電解液進行了慢掃速下的窗口測試,以及正負極(LiMn2O4正極以及C-TiO2負極)在HCS-ACE中的半電池測試。
我們將HCS-ACE應用在全電池中,首先進行了恒壓充電觀測電流衰減,評估電池的自放電性能等,原位壓力測試等手段充分驗證了電池的性能。在保證電池綜合性能良好后,將電池放大成為mAh級別的單層軟包電池,相較于ACE電解液,使用HCS-ACE電解液的全電池具有更高的首周效率(圖5b),具有更加優異的循環性能(圖5c),更為重要的是,單層軟包電池的能量密度能夠達到40Wh/kg。當全電池使用3C循環時,也能保持良好的循環性能(圖5d-e)。
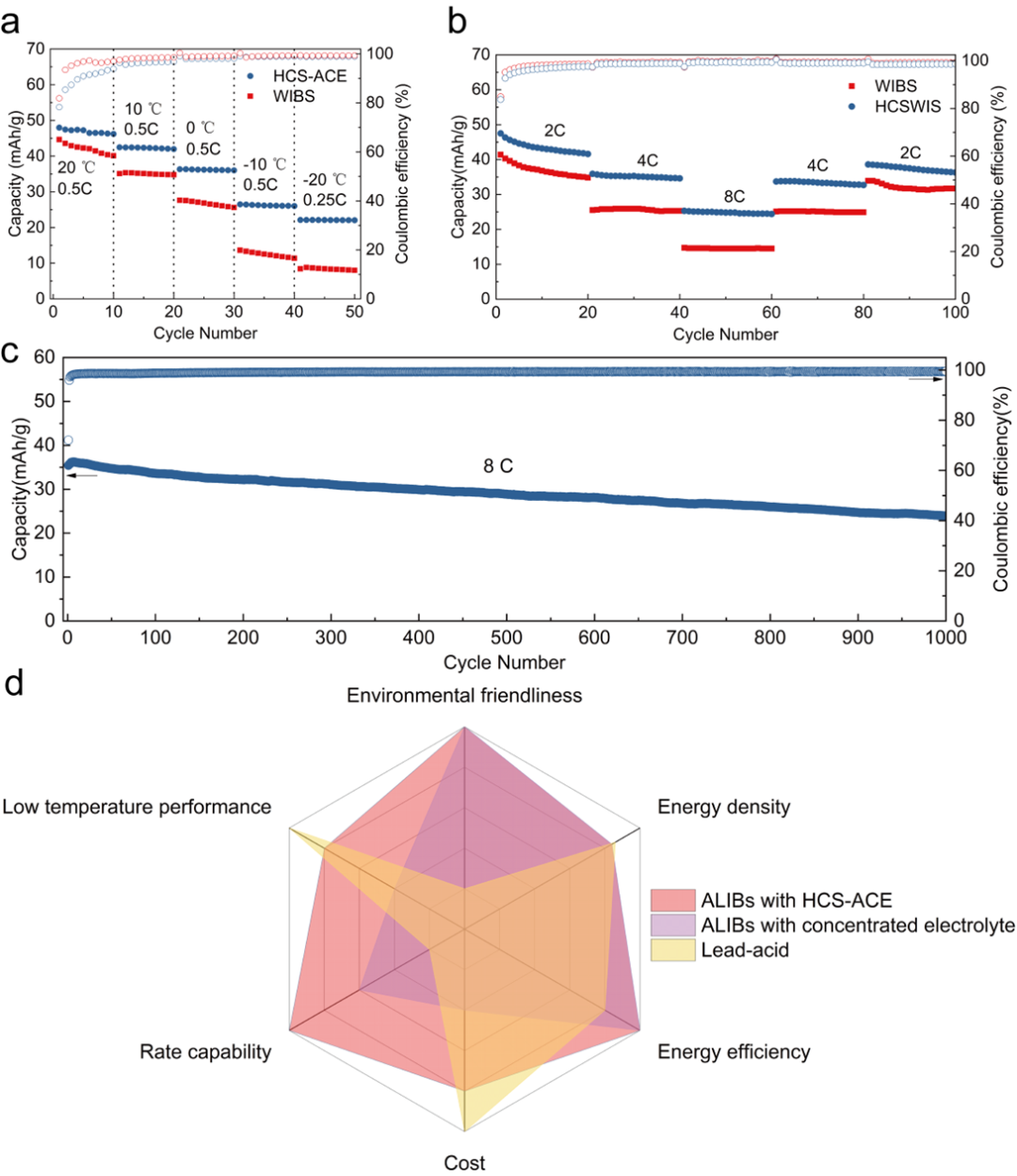
圖6.HCS-ACE的倍率性能和低溫性能。a)HCS-ACE與WIBS的全電池低溫性能對比。b))HCS-ACE與WIBS的倍率性能對比c)HCS-ACE全電池8C長循環性能。d)高鹽濃度鋰離子電池,HCS-ACE鋰離子電池與鉛酸電池的綜合性能比較雷達圖。
相比于高鹽濃度WIBS的全電池,使用HCS-ACE的全電池擁有更好的低溫性能(圖6a),更好的倍率性能(圖6b),良好的高倍率長循環性能(圖6c)。最后對高鹽濃度鋰離子電池,HCS-ACE鋰離子電池以及鉛酸電池的綜合性能進行了比較,相較于高鹽濃度鋰離子電池,擁有HCS-ACE的水系鋰離子電池具有更好的倍率性能,更低的成本以及更好的低溫性能。相比于鉛酸電池,則具有更好的倍率性能,更好的環境友好性已經更高的能量效率。
本文證明了界面調控對于水系電池的重要性,如能夠對界面結構進行合理的調整,在水系電池中高鹽濃度的電解液可能不是必要的條件。更為重要的是,本文提出了降低電解液濃度的策略,結合低成本正負極材料的使用,為水系電池服務大規模儲能提供新的可行途徑。

馮光教授:華中科技大學能源與動力工程學院教授/博導,英國皇家化學學會會士,研究方向聚焦于微納尺度界面和輸運相關的基礎問題。研究工作具有能源與物理、化學、材料等方向相交叉的學科特點;已發表英文書籍3章、SCI期刊論文百余篇,其中第一/通訊作者論文60多篇(包括NatureMaterials、NatureComputational Science、NatureCommunications、PhysicalReview X、AdvancedMaterials等),H因子37。現任《EnergyAdvances》創刊副主編、SCI期刊《ChemElectroChem》、《FluidPhase Equilibria》的編委和《GreenEnergy & Environment》的青年編委。

索鎏敏研究員:研究員,博士生導師。2013年于中國科學院物理研究所獲理學博士學位,曾先后在在美國馬里蘭大學,美國麻省理工學院從事博士后研究工作,2017年加入中國科學院物理研究所。近年來發表SCI論文共計 70篇(IF>10, 57篇),申請發表專利 25 項。通訊/一作身份發表研究類論文 42 篇,包括Science、NatureEnergy/Nature Chemistry(2篇)、Nat.Commun./Sci. Adv./PNAS(3篇)、Adv.Mater/Angew/JACS(11 篇)、Matter、Adv.Energy.Mater/ACSEnergy Letter/Energy Storage Material (10 篇)、ACSNano/Nano Letter (3 篇)等。文章發表以來SCI引用次數大于 10000 次,其中 98%源于研究類論文貢獻,60%以上源于通訊/第一作者論文貢獻,引用次數:>1000次(2篇),H因子43。
Zhou,A#.,Zhang, J#.,Chen, M., Yue, J., Lv, T., Liu, B., Zhu, X., Qin, K., Feng, G*.and Suo, L*.,Electric-field-reinforced Hydrophobic Cationic Sieve Lowers theConcentration Threshold of Water-in-Salt Electrolytes. Adv.Mater. (2022) https://doi.org/10.1002/adma.202207040